
James Collins团队开发用于生物学研究的超高速人工智能系统,已开源代码
来源:生辉 2023-07-17 11:02
过去几十年,生物数据集的规模与复杂性大幅增长,利用机器学习为潜在生物过程构建信息与预测模型能够加速生物工程和生物医学应用的进展。
过去几十年,生物数据集的规模与复杂性大幅增长,利用机器学习为潜在生物过程构建信息与预测模型能够加速生物工程和生物医学应用的进展。对于难以预测的生物系统的行为,机器学习技术能够提供所需的预测能力,而不需要详细的机械理解。
在生物学中使用机器学习有两个目标。一是在缺乏实验数据的地方做出准确的预测,并利用这些预测来指导未来的研究工作;二是使用机器学习来进一步了解生物学。
对于不同类型的生物数据,需要选择特定的机器学习技术,为其相匹配最适合的模型,这一过程往往要耗时几周。根据 Google 机器学习基础课程,仅数据准备和转换就占用了项目时间的 80%。
同时,这也是一项成本高昂的工作,研究者通常必须投资于重要的数字基础设施和训练有素的人力资源,然后才能确定他们的想法是否可行。
此外,大多数 AutoML 工具只能探索和构建简化类型的模型。
如何让生物学和机器学习之间更有效地合作?甚至没有机器学习专业知识也可以构建机器学习模型呢?
在一项开创性的研究中,麻省理工学院 (MIT) 的研究人员开发了一种名为 BioAutoMATED 的自动化机器学习系统,可以生成用于生物学研究的人工智能模型。相关文章以题“BioAutoMATED: An end-to-end automated machine learning tool for explanation and design of biological sequences”发表于 Cell Systems 杂志。

(来源:Cell Systems)
该团队由 Termeer 医学工程和科学教授 James J. Collins 领导,旨在简化生物学领域科学家和工程师构建机器学习模型的过程。这个创新系统不仅为给定的数据集选择和构建适当的模型,甚至可以处理繁重的数据预处理任务,将长达数月的过程缩短到几个小时。
剑桥大学的首席研究员 Katie Collins 强调了 BioAutoMATED 的这种多功能性,并表示:“它使用户能够在单一框架内识别模式、提出更好的问题并从生物数据中快速获得答案,而无需广泛的 ML 专业知识。”
节省时间,加速序列的分析和设计
生物学的基本语言是基于序列的。生物序列,如 DNA、RNA、蛋白质和聚糖,具有惊人的信息属性,即在本质上是标准化的,就像一个字母表。很多 AutoML 工具主要专注于图像和文本识别,但 BioAutoMATED 将 AutoML 的功能扩展到生物序列。
为了构建 BioAutoMATED ,该团队修改了三个现有的 AutoML 工具,每个工具都使用不同的方法来生成模型:AutoKeras,用于搜索最佳神经网络;DeepSwarm,使用基于群的算法来搜索卷积神经网络(convolutional neural networks);TPOT,它使用包括遗传编程和自学习在内的多种方法来搜索非神经网络。BioAutoMATED 为所有三种工具生成标准化输出结果,以便用户可以轻松比较它们并确定哪种类型可以从其数据中产生最有用的见解。
根据资料,BioAutoMATED 能够将任何长度、类型或生物功能的 DNA、RNA、氨基酸和聚糖序列作为输入。BioAutoMATED 自动预处理输入数据,然后生成可以仅根据序列信息预测生物功能的模型。
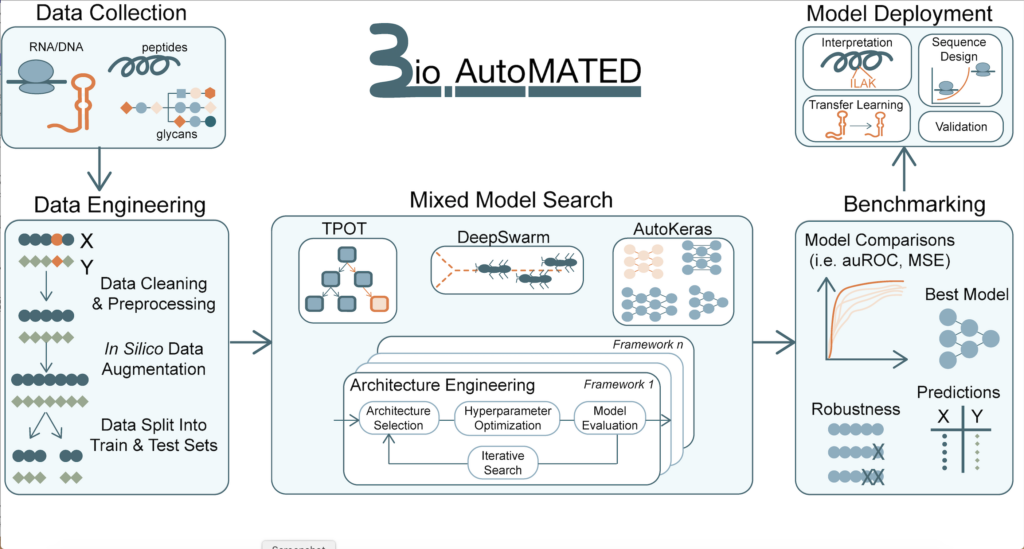
▲图丨BioAutoMATED 是一种集成的 AutoML 工具。(来源:Wyss Institute at Harvard University)
之后,为了探索该平台的功能,背后的研究人员改变核糖体结合位点序列,观察其对大肠杆菌中核糖体结合效率的影响。结果很惊喜,因为 BioAutoMATED 成功识别了 DeepSwarm 算法生成的模型,可以准确预测翻译效率。
值得注意的是,该模型的性能与专业机器学习专家创建的模型相当,同时只需要一小部分时间和最少的代码输入。此外,BioAutoMATED 提供了对决定翻译效率至关重要的序列特征的见解,甚至促进了用于实验测试的新序列的设计。
研究人员进一步探索发现,该平台擅长识别肽序列中影响抗体与药物结合的关键氨基酸,并根据其序列有效地将不同类型的聚糖分为免疫原性组和非免疫原性组。
优势广泛,提供更大的搜索空间
BioAutoMATED 专为几乎没有 ML 经验的生物学家设计,它的主要优势之一是它能够探索和构建各种类型的监督机器学习模型。其中包括二元分类模型、多类分类模型和回归模型。通过将多个工具整合到一个框架内,BioAutoMATE 提供了比单个 AutoML 工具更大的搜索空间,从而使模型选择更加灵活和准确。
其次,BioAutoMATED 对于拥有较小、稀疏生物数据集的研究小组特别有利,它可以探索更适合此类数据集的模型以及更复杂的神经网络。这样一来,即使在数据不十分充足的情况下,研究人员可以充分利用可用数据并获得有意义的见解。
另一方面,BioAutoMATED 甚至能够帮助确定需要多少数据来适当地训练所选模型。
借助 BioAutoMATED,研究人员可以运行初始实验,以评估是否值得聘请机器学习专家来建立不同的模型进行进一步的实验。通常来说,聘请机器学习专家需要一笔不小的资金,这样能够帮助研究人员避免不必要的经济损失。
虽然 BioAutoMATED 生成的所有模型都应该经过实验验证,但研究人员设想将其集成到不断扩展的 AutoML 工具库中,从而有可能将其应用范围从生物序列扩展到其他类似序列的对象,例如指纹。
为了促进协作和可访问性,研究人员在 GitHub 上公开了 BioAutoMATED 的代码,并鼓励其他人利用和改进代码,促进科学界内的合作。研究人员设想,未来 BioAutoMATED 将成为所有人都可以使用的有价值的工具,将严格的生物学实践与快速发展的 AI-ML 技术相结合。
BioAutoMATED 的开发代表了生物学研究领域的重大突破。通过简化模型选择和数据预处理,这一创新系统简化了研究流程,并降低了生物科学研究人员的进入壁垒。随着该领域的不断发展,协作和发现的可能性是无限的。
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
