
Genome Biology:开发出单细胞基因组单分子测序新方法
来源:生物医学前沿创新中心 2021-07-07 20:35
单细胞全基因组测序技术(scWGS)可以有效揭示生物样品中不同细胞之间的异质性,并系统鉴定单个细胞的基因组中发生的遗传变化,例如拷贝数变异(CNV)和点突变(单核苷酸变异,SNV)等。过去十年,研究人员已经开发出多种单细胞基因组扩增技术,例如简并寡核苷酸引物PCR扩增技术(DOP-PCR)、多重置换扩增技术(MDA)、多重退火和基于环的扩增循环技术(MALB
单细胞全基因组测序技术(scWGS)可以有效揭示生物样品中不同细胞之间的异质性,并系统鉴定单个细胞的基因组中发生的遗传变化,例如拷贝数变异(CNV)和点突变(单核苷酸变异,SNV)等。过去十年,研究人员已经开发出多种单细胞基因组扩增技术,例如简并寡核苷酸引物PCR扩增技术(DOP-PCR)、多重置换扩增技术(MDA)、多重退火和基于环的扩增循环技术(MALBAC),以及通过转座子插入和体外转录进行线性扩增技术(LIANTI)等。但是,目前的单细胞全基因组测序技术均基于二代测序(NGS)平台,该平台检测准确度高,但是测序读长相对较短(通常只有150bpX2),主要适用于检测单个细胞中的单核苷酸变异(SNV)、小的插入缺失(<50bp,Indel)以及拷贝数变异(CNV)等。由于二代测序平台读长的限制,对于基因组中结构变异的检测具有很大的局限性。结构变异(包括插入、缺失、重复和易位等)是人类体细胞遗传变异的主要来源之一,对肿瘤的发生、发展和转移具有潜在的驱动作用,然而目前在单个细胞水平对基因组结构变异的研究却鲜有报道。
针对基于二代测序平台的单细胞基因组测序技术难以高效鉴定单个细胞中结构变异这一世界难题,北京未来基因诊断高精尖创新中心、北京大学生物医学前沿创新中心汤富酬课题组在Genome Biology上在线发表了题为“SMOOTH-seq: single-cell genome sequencing of human cells on athird-generation sequencing platform”的研究论文,在国际上率先开发了基于三代测序(单分子测序)平台的单细胞基因组测序技术。
该研究的主要突破有:
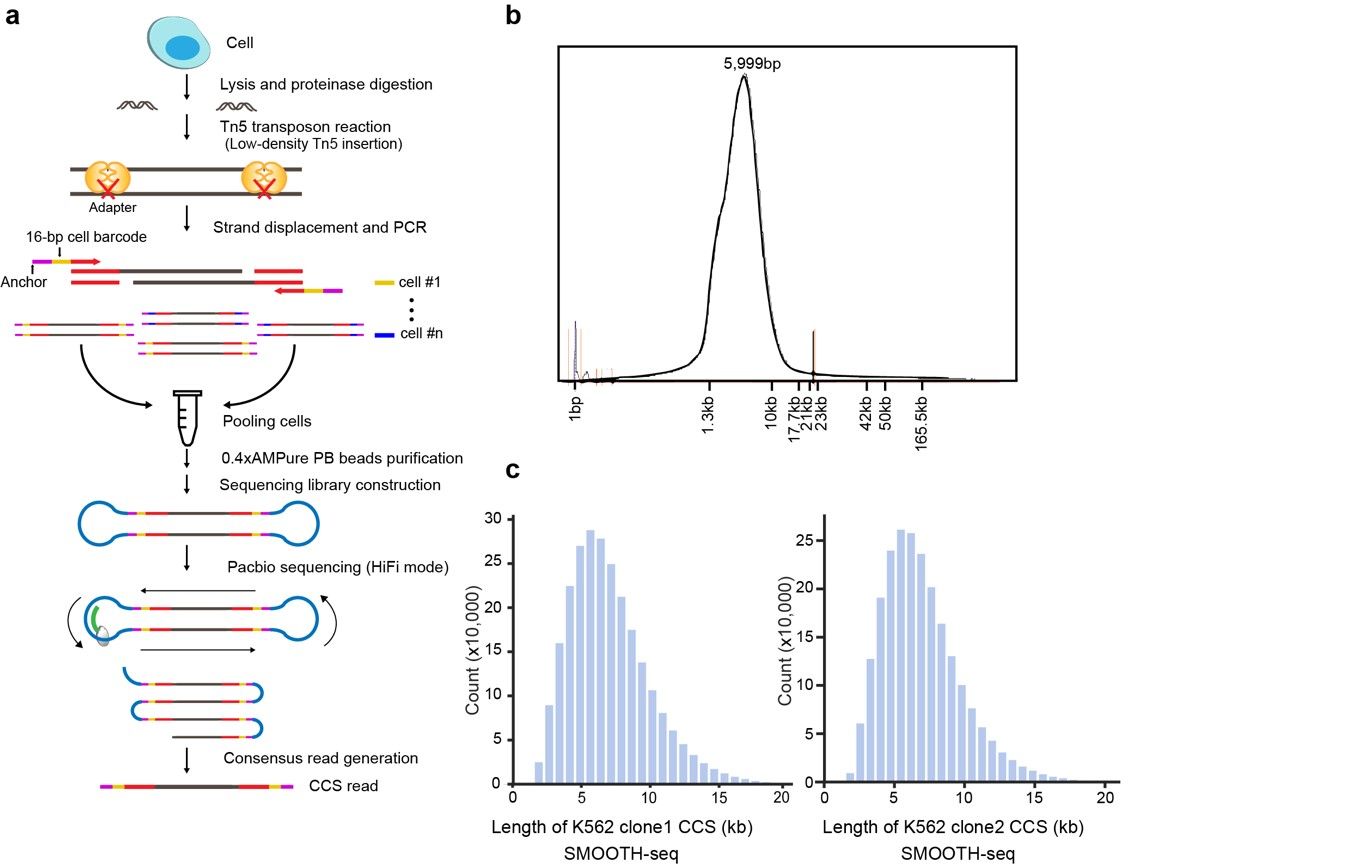
开发了一种高精度的基于三代测序(单分子测序)平台的单细胞基因组测序方法——SMOOTH-seq(Single-MOlecule real-time sequencing of LOng Fragments amplified THrough Transposon insertion)。使用优化后的Tn5转座反应,SMOOTH-seq能够从单个细胞中扩增出平均长度约6kb的基因组片段(测序读长比单细胞基因组二代测序技术长了20倍左右),通过引入与单分子测序平台兼容的细胞条形码使单细胞基因组DNA扩增子适用于Pacbio sequel II平台的HiFi测序模式。测序后的数据中,产生的环化测序(circular consensus sequencing, CCS)的读长平均在6kb左右, 最长可达43kb。
该研究开发的SMOOTH-seq方法能够在单个细胞中高效检测基因组结构变异。在单个K562细胞中,当测序深度仅为0.4X时,基因组覆盖度可达19%。该研究对91个单细胞进行SMOOTH-seq分析,从中检测到4,790 个缺失事件和5,589个插入事件,其中87%的缺失片段和91% 的插入片段长度小于1kb,检测到的插入事件DNA片段最长达到7.7kb。同时,该研究也在K562细胞中检测到521个易位事件,包括准确检测到两对经典融合基因:BCR-ABL 和NUP214-XKR3。SMOOTH-seq技术对结构变异的检测精度高,当使用K562大量细胞的基因组三代测序结果作为比较基准时,该研究中使用的K562细胞系的每个单细胞中的结构变异检测平均精确度为75%,特别是在单个细胞中检测插入事件平均精确度为85%。此外,SMOOTH-seq 也可以以1Mb 的分辨率准确检测到两个K562克隆之间的不同拷贝数变异(CNV)事件。
该研究开发的SMOOTH-seq方法能够在单个细胞中高效检测染色体外环形DNA(ecDNA)。已有的研究报道表明,染色体外环形DNA在肿瘤发生中比较常见,且致癌基因能够在染色体外环形DNA中进行大量扩增,促进肿瘤发生和转移。SMOOTH-seq技术产生的长读长数据,使其能够被用于在单个细胞中精准捕获小于10kb的全长染色体外环形DNA。本研究同时开发了用于鉴定K562细胞系中的染色体外环形DNA的生物信息学分析方法。当仅有一个拷贝的Tn5转座酶与一个染色体外环形DNA分子结合时,整个环形DNA分子就可被完整扩增为一个线性片段,即单个读段即可覆盖一个染色体外环形DNA分子的全长序列。通过统计Tn5插入位置不同但长度完全相同的一组读段,即可判断它们是否来源于同一个环形DNA。同时该特征可用于帮助精准区分染色体外环形DNA和串联重复序列(如图3所示)。
该研究开发的SMOOTH-seq方法能以较高准确度检测基因点突变(SNV)。
该研究开发的SMOOTH-seq方法可以在结直肠癌肿瘤样本中准确检测出各种基因组结构变异事件。在对患者结直肠癌肿瘤样本的分析中,以在结直肠癌的至少2个单细胞中同时检测到为标准,该研究检测出8,594个结构变异事件(4,089个插入事件,3,852个缺失事件,341个易位事件,以及312个重复事件)。通过将结直肠癌肿瘤样本和K562细胞系中共有的结构变异去除后,该研究共得到3,570个结直肠癌肿瘤细胞特异性的结构变异事件(1,376 插入事件, 1,661 缺失事件, 230 易位事件以及303 重复事件)。同时,该研究通过设置多个对照基因组(包括相应的肿瘤组织、与肿瘤相邻的正常组织、GM12878细胞系和另一个体的外周血单核细胞的基因组)对检测出的结构变异事件进行了PCR验证。此外,该研究发现结直肠癌肿瘤样本和K562细胞系中共有的结构变异在所有被检测的多个人类基因组DNA样品中均存在,说明这些结构变异事件实际上是由于当前的人类参考基因组不够完善,缺失了部分关键序列信息引起的。今后三代测序将有助于组装出更完整精准的人类参考基因组序列(生物谷 bioon)
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->
