
Genome Biology:科研人员开发出在单细胞中识别染色质类染色质拓扑相关结构域结构的算法
来源:北京基因组所 2021-08-02 08:38
基因组DNA和组蛋白以特定的形式高度折叠在细胞核中,这一高级结构即三维基因组学,对细胞核内的诸多生命活动至关重要。基于染色质构象捕获(3C),尤其是高通量技术(Hi-C,ChIA-PET)的发展推动了三维基因组的研究,发现了包括染色质拓扑相关结构域(TAD),染色质环等一系列层次化的结构特征。近年来,单细胞水平下的Hi-C研究成为三维
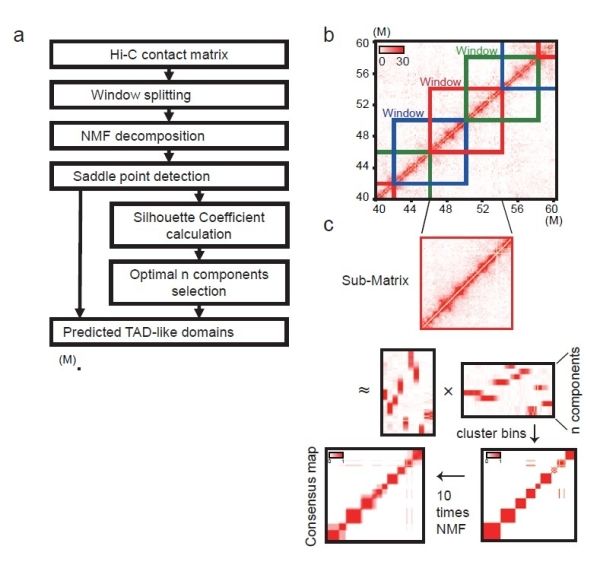
基因组DNA和组蛋白以特定的形式高度折叠在细胞核中,这一高级结构即三维基因组学,对细胞核内的诸多生命活动至关重要。基于染色质构象捕获(3C),尤其是高通量技术(Hi-C,ChIA-PET)的发展推动了三维基因组的研究,发现了包括染色质拓扑相关结构域(TAD),染色质环等一系列层次化的结构特征。近年来,单细胞水平下的Hi-C研究成为三维基因组的重要研究方向。单细胞Hi-C数据对深入理解染色质结构的动力学机制,建立高分辨率细胞发育图谱都具有重要意义。然而,单细胞Hi-C数据由于极度稀疏,目前主流的Hi-C数据分析算法对其无能为力,而针对单细胞Hi-C设计的算法亦表现不佳。因此,亟须新的计算方法来分析鉴定单细胞内的染色质高级结构。
中国科学院北京基因组研究所(国家生物信息中心)张治华研究组开发了预测单细胞内类TAD结构的算法deTOKI。相关研究成果以DeTOKI identifies and characterizes the dynamics of chromatin TAD-like domains in a single cell为题,发表在Genome Biology上。
该研究将deTOKI与适用于低分辨率水平Hi-C数据的新算法IS、deDoc、SpectralTAD、GRiNCH,以及先由单细胞Hi-C实验数据通过预测出高分辨率数据,再由已有算法鉴定类TAD域结构的新算法scHiCluster及Higashi六个软件进行综合比较,发现用deTOKI分析单细胞Hi-C数据结果优于其他六个软件。研究比较的内容主要基于两点,首先是将高分辨率水平的Hi-C数据进行下采样,比较下采样数据和原始数据中鉴定的类TAD域结构的相似度,然后对染色质结构进行三维建模,对各个模型分别生成高分辨率水平和单细胞水平的模拟Hi-C数据,比较两个数据中鉴定的类TAD域结构的相似度。该研究还在已有的单细胞Hi-C实验数据上使用模块系数和结构熵等指标来评价软件的表现,deTOKI均优于其他算法。
新算法deTOKI有助于未来的单细胞内染色质高级结构的研究,基于deTOKI算法,研究发现了单细胞内的类TAD域结构和细胞类型的关系,以及其与组蛋白修饰、DNA甲基化等多组学数据的关联。该研究丰富了关于基因组结构和功能关系的认识,为三维基因组学研究提供了新思路。(生物谷Bioon.com)
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
