
Nature 子刊:多组学数据与机器学习识别新的癌症基因
来源:本站原创 2021-04-30 14:41
癌症基因的鉴定在精准肿瘤学和癌症治疗学的发展中起着至关重要的作用。癌症的进展被认为是由驱动基因突变的积累引起的,这些突变赋予细胞选择性的生长优势。在过去的几年里,人们致力于从遗传大数据中找出癌症基因,预测癌症基因的最初方法是寻找显着突变的基因,然而,目前已知的癌症基因(KCG)目录的完整性仍存在争议。尽管成千上万的癌症基因组序列有助于癌症基因的鉴定,但仍存在
癌症基因的鉴定在精准肿瘤学和癌症治疗学的发展中起着至关重要的作用。癌症的进展被认为是由驱动基因突变的积累引起的,这些突变赋予细胞选择性的生长优势。在过去的几年里,人们致力于从遗传大数据中找出癌症基因,预测癌症基因的最初方法是寻找显着突变的基因,然而,目前已知的癌症基因(KCG)目录的完整性仍存在争议。
尽管成千上万的癌症基因组序列有助于癌症基因的鉴定,但仍存在以下挑战:第一,在某些肿瘤类型中已鉴定的癌症基因数量仍然很低;第二,许多在肿瘤发生中起重要作用的基因在其DNA序列水平上没有改变,但通过诸如DNA甲基化、低甲基化和拷贝数改变等各种细胞机制失调。这类未突变的癌症依赖性基因引起了人们极大的兴趣,因为其中许多是转录和表观遗传调节因子,可用于小分子治疗的靶向治疗。此外,基因在信号和调控途径以及蛋白质复合物中共同作用。
为了利用多组学数据中包含的补充信息,需要开发能够表示不同数据层并将其集成到单个框架中的模型。生物网络可以被视为图形,其中节点代表基因,节点之间的连接代表基因-基因相互作用,而组学数据水平可以被视为基因的特征向量。最近的一些计算方法侧重于通过同时整合不同类型的基因组数据来预测癌症基因或识别癌症基因模块,很少有方法将多维节点向量与基因-基因相互作用的图形表示结合起来。在过去几年中,深度学习模式在分子生物学和基因组学领域取得了前所未有的成果。图形深度学习将图形结构纳入深度学习框架,图卷积网络(GCN)能够基于网络中的未标记节点的相关特征向量以及网络的拓扑对其进行分类,使得能够以自然的方式将基于图的数据与特征向量集成。
2021年4月12日,德国慕尼黑环境卫生研究中心计算生物学研究所的研究团队在《Nature Machine Intelligence》期刊发表题为“Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms”的研究论文。

该研究团队开发了一种基于图卷积网络的机器学习方法: EMOGI,通过将突变、拷贝数变化、DNA甲基化和基因表达等多组学泛癌数据与蛋白质-蛋白质相互作用(PPI)网络相结合来预测癌症基因。该研究团队预测了165个新的癌症基因,这些基因与PPI网络中已知的癌症驱动因素相互作用,而不是自身高度突变,新的预测还丰富了通过功能丧失筛查确定的必需基因。该方法能够找到由不同的分子改变而非高突变率定义的癌症基因类别,阐释了基因如何促进或阻碍肿瘤的发展和进展。
该研究团队开发的EMOGI以图卷积网络(GCNs)为基础,以半监督的方式进行训练,以区分假定的癌症和非癌症基因。通过EMOGI分析来自TCGA的16种癌症类型的基因组数据,输出了一个完全标记的图,其中每个基因都被分配了成为癌症基因的概率。
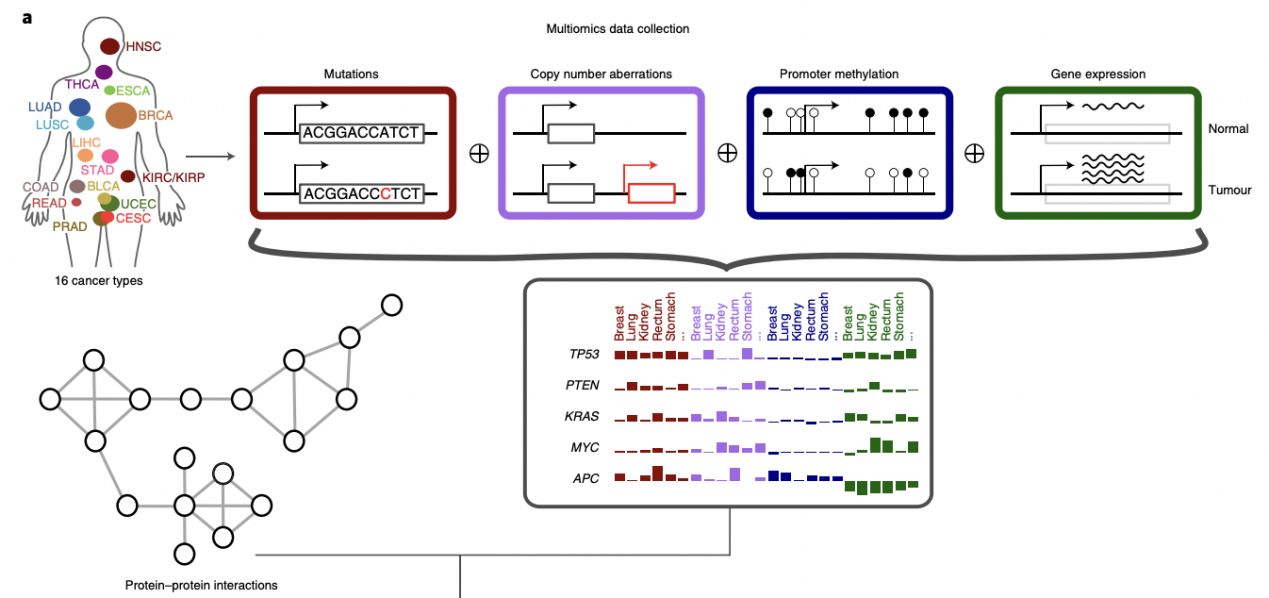
EMOGI框架的示意图
接下来研究团队使用分层相关传播(LRP)进行特征重要性分析来解释由EMOGI鉴定的四个癌症基因(APC,TWIST1,STIL和MYC)的分子特征,还使用LRP规则提取了来自PPI网络的相互作用对单个基因分类的贡献,发现突变频率是癌症基因分类最重要的特征,尤其是对于最重要的预测。
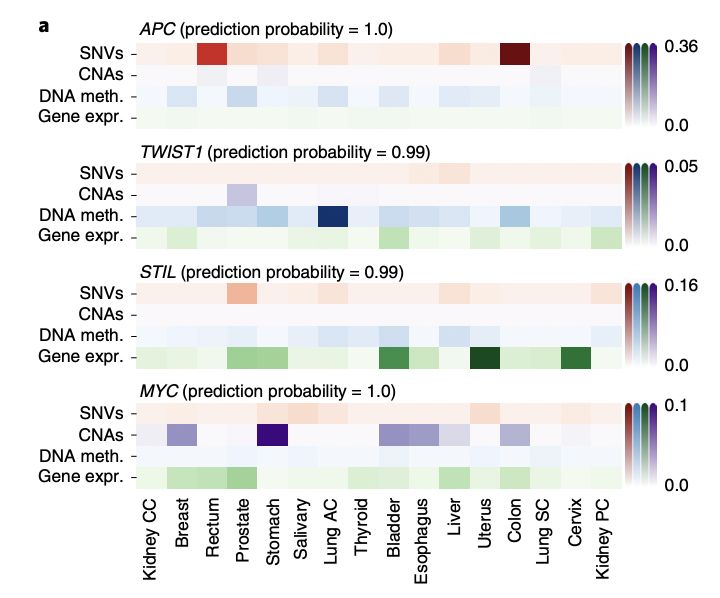
几种癌症基因模型的解释概括了其致癌分子机制
使用Kluger的光谱双聚类算法,根据不同癌症类型的特征重要性LRP得分,从共识路径数据库(CPDB)网络中对EMOGI的前1000个预测癌症基因进行聚类,揭示了具有独特功能特征的不同类别的癌症基因。

特征贡献的聚类揭示了不同类别的癌症基因
最后,结合网络中所有基因的信息,研究团队建立了基因-基因LRP贡献的有向加权图,并研究图的强连接节点(SCC)。SCC中包含来自CPDB网络的323个基因。研究团队提取了45个模块,这些模块包含两个以上的基因,并确定了165个新的癌症基因,这些基因与PPI网络中已知的癌症驱动因素相互作用。
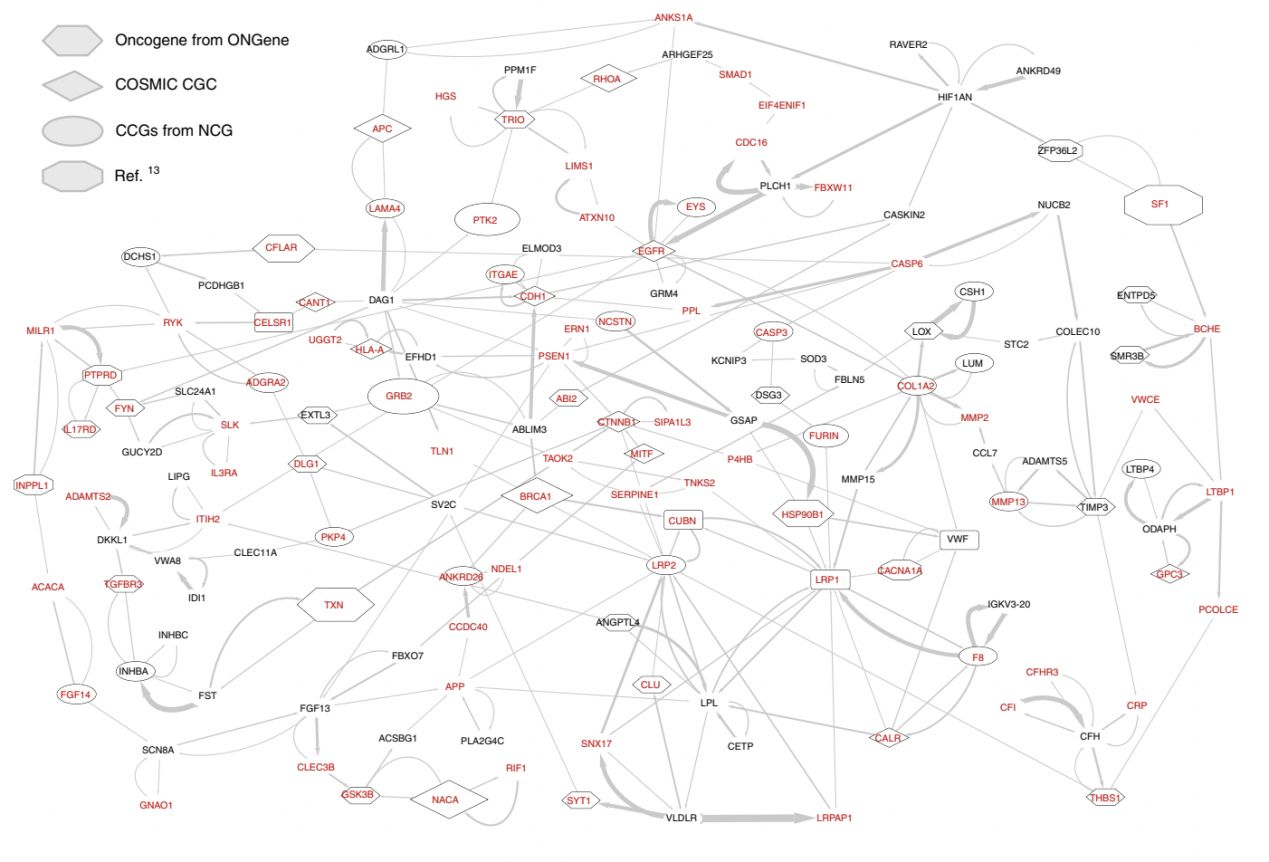
EMOGI可提取与癌症基因分类重要的子网络相对应的PPI网络节点
总而言之,该研究团队基于图卷积网络开发了一种用于可解释的多组学图集成(EMOGI)的机器学习方法,以对来自大型数据集的癌症基因进行优先排序,预测了165个新的癌症基因,它们与PPI网络中的已知癌症驱动程序相互作用。通过应用EMOGI,能够找到由除高突变率以外的不同分子改变所定义的癌症基因类别,从而有助于理解基因如何影响肿瘤。
原文出处:
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->
