
Mol Cancer:抑制V-ATPase可降低去势抵抗前列腺癌中突变雄激素受体活性
来源:本站原创 2021-04-24 20:04
近期,Bradleigh Whitton教授及其团队开展了有关V-ATPase和前列腺癌关系的实验,实验结果表明V-ATPase失调通过影响AR功能直接与激素反应性前列腺癌和CRPC有关。特别是,V-ATPase抑制可以减少AR信号传导,而与突变AR的表达无关。
2021年4月24日讯/生物谷BIOON/临床上大多数晚期前列腺癌患者随后发展为具有侵袭性的去势抵抗前列腺癌(CRPC)表型,通常与剪接变异体或突变AR形式的表达有关。尽管目前的证据表明,空泡ATPase(V-ATPase)是一种催化质子跨细胞膜和质膜转运的多蛋白复合物,可以影响野生型AR功能,但V-ATPase抑制对变异AR功能的影响尚不清楚。
近期,Bradleigh Whitton教授及其团队开展了有关V-ATPase和前列腺癌关系的实验,实验结果表明V-ATPase失调通过影响AR功能直接与激素反应性前列腺癌和CRPC有关。特别是,V-ATPase抑制可以减少AR信号传导,而与突变AR的表达无关。
目前有限的数据表明V-ATPase在前列腺癌中有可能发挥一定的作用,特别是在AR调节方面。V-ATPase的表达与前列腺癌细胞的侵袭性和转移潜能相关,V-ATPase调节蛋白如LASS2/TMSG1和PEDF可以通过调节V-ATPase活性来影响肿瘤细胞的生长和侵袭。HSPC (LNCaP)和CRPC (C4-2B)细胞系中V-ATPase的化学抑制降低了两种细胞系的体外侵袭能力。V-ATPase抑制导致PSA重新定位溶酶体样胞内囊泡。此外,LNCaP和LAPC4细胞中V-ATPase的抑制同时耗尽了AR蛋白和mRNA。
以上研究结论支持了在前列腺癌中AR和V-ATPase功能之间的潜在联系。该实验正是来探索这种相互作用,包括与临床向CRPC过渡相关的AR形式的改变。
研究人员首先测定了HSPC LNCaP细胞系、HEK-293和雄激素不敏感22Rv1细胞系中V-ATPase抑制剂baff - a1和con-A的非细胞毒性浓度。然后使用不表达AR的HEK-293细胞系,测量V-ATPase抑制对野生型AR (AR- wt) transactivation的影响。用ARE报告质粒、AR- wt表达质粒和Renilla转染质粒转染HEK-293细胞。在1 nmol/L DHT和不同浓度的V-ATPase抑制剂baf- a1或con-A暴露24小时后,通过发光定量检测ARE基因激活水平,再对其表达蛋白质进行印迹分析。
综上所述,V-ATPase抑制可降低前列腺癌细胞AR信号转导。最重要的是,这是首次证明延伸到点突变和剪接变异的AR形式与向CRPC的转化临床相关。这表明V-ATPase可以作为一种克服AR异常患者的AR信号途径,包括未满足临床需求的CRPC患者。
近期,Bradleigh Whitton教授及其团队开展了有关V-ATPase和前列腺癌关系的实验,实验结果表明V-ATPase失调通过影响AR功能直接与激素反应性前列腺癌和CRPC有关。特别是,V-ATPase抑制可以减少AR信号传导,而与突变AR的表达无关。

2018年,128万男性被诊断患有前列腺癌,35.9万人死亡。最初,前列腺癌细胞的生长依赖于雄激素受体(AR)的激活。因此,目前晚期患者多采用永久性雄激素剥夺疗法(ADT)治疗以限制癌细胞生长。然而,从激素敏感前列腺癌(HSPC)也有可能因此发展为更具侵袭性的去势抵抗前列腺癌(CRPC)表型。但CRPC的预后很差,中位生存期约为2年。CRPC的预后部分归因于通过AR自身的适应性改变而持续的AR信号。
目前有限的数据表明V-ATPase在前列腺癌中有可能发挥一定的作用,特别是在AR调节方面。V-ATPase的表达与前列腺癌细胞的侵袭性和转移潜能相关,V-ATPase调节蛋白如LASS2/TMSG1和PEDF可以通过调节V-ATPase活性来影响肿瘤细胞的生长和侵袭。HSPC (LNCaP)和CRPC (C4-2B)细胞系中V-ATPase的化学抑制降低了两种细胞系的体外侵袭能力。V-ATPase抑制导致PSA重新定位溶酶体样胞内囊泡。此外,LNCaP和LAPC4细胞中V-ATPase的抑制同时耗尽了AR蛋白和mRNA。
以上研究结论支持了在前列腺癌中AR和V-ATPase功能之间的潜在联系。该实验正是来探索这种相互作用,包括与临床向CRPC过渡相关的AR形式的改变。
研究人员首先测定了HSPC LNCaP细胞系、HEK-293和雄激素不敏感22Rv1细胞系中V-ATPase抑制剂baff - a1和con-A的非细胞毒性浓度。然后使用不表达AR的HEK-293细胞系,测量V-ATPase抑制对野生型AR (AR- wt) transactivation的影响。用ARE报告质粒、AR- wt表达质粒和Renilla转染质粒转染HEK-293细胞。在1 nmol/L DHT和不同浓度的V-ATPase抑制剂baf- a1或con-A暴露24小时后,通过发光定量检测ARE基因激活水平,再对其表达蛋白质进行印迹分析。
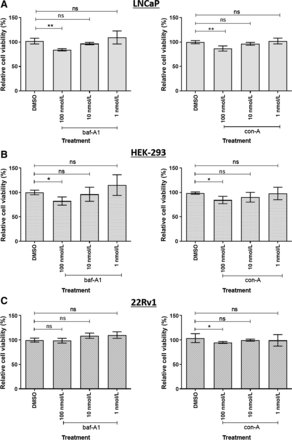
MTS法测定baf-A1和con-A对细胞系的毒性反应
与之前的研究一致,baf-A1和con-A的浓度显著降低了AR-WT活性。在蛋白水平上,V-ATPase抑制也降低了AR表达,这与LNCaP和LAPC4 HSPC细胞中先前的数据一致。
值得注意的是,尽管减少AR蛋白的表达是一个一致的发现,我们发现V-ATPase抑制对PSA蛋白的表达有不同的影响。当V-ATPase抑制与AR拮抗联合作用时,AR下游转录本表达和AR蛋白表达均显著降低。
我们已经证明,V-ATPase抑制降低了AR-V7、AR-Q641X和AR-F877L的活化。此外,V-ATPase抑制导致22Rv1细胞和表达诱导型F877L/T878A双突变体的LNCaP细胞AR蛋白表达减少。总之,这些数据加强了V-ATPase和AR信号之间联系的证据。
值得注意的是,尽管减少AR蛋白的表达是一个一致的发现,我们发现V-ATPase抑制对PSA蛋白的表达有不同的影响。当V-ATPase抑制与AR拮抗联合作用时,AR下游转录本表达和AR蛋白表达均显著降低。
我们已经证明,V-ATPase抑制降低了AR-V7、AR-Q641X和AR-F877L的活化。此外,V-ATPase抑制导致22Rv1细胞和表达诱导型F877L/T878A双突变体的LNCaP细胞AR蛋白表达减少。总之,这些数据加强了V-ATPase和AR信号之间联系的证据。
综上所述,V-ATPase抑制可降低前列腺癌细胞AR信号转导。最重要的是,这是首次证明延伸到点突变和剪接变异的AR形式与向CRPC的转化临床相关。这表明V-ATPase可以作为一种克服AR异常患者的AR信号途径,包括未满足临床需求的CRPC患者。
原始出处:Bradleigh Whitton,Haruko Okamoto,et
al.V-ATPase Inhibition Decreases Mutant Androgen Receptor Activity in
Castrate-resistant Prostate Cancer. MOL CANCER April 2021 DOI:10.1158/1535-7163.MCT-20-0662
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->
