
如何理解阿尔茨海默病新药的详细数据
来源:药明康德 2019-12-07 17:06
今年10月23日,渤健(Biogen)和卫材(Eisai)公司联合宣布,计划在明年向FDA递交β淀粉样蛋白(β-amyloid)抗体aducanumab的生物制品申请(BLA),治疗早期阿尔茨海默病(AD)患者。而在今年3月,渤健和卫材曾经宣布,由于独立数据监督委员会认为aducanumab达到预期疗效的可能性很小,终止aducanumab的3期临床试验。这
今年10月23日,渤健(Biogen)和卫材(Eisai)公司联合宣布,计划在明年向FDA递交β淀粉样蛋白(β-amyloid)抗体aducanumab的生物制品申请(BLA),治疗早期阿尔茨海默病(AD)患者。而在今年3月,渤健和卫材曾经宣布,由于独立数据监督委员会认为aducanumab达到预期疗效的可能性很小,终止aducanumab的3期临床试验。这一“惊天逆转”的消息一出,可谓“一石激起千层浪”,引起了业界和大众的广泛热议。自2003年以来,美国FDA尚未批准任何AD新药,aducanumab的“起死回生”无疑为成千上万的AD患者带来了一线曙光。
渤健在10月23日召开的电话会议上公布了对aducanumab在两项3期临床试验(EMERGE和ENGAGE)中试验数据的分析结果,然而这些数据让业内专家非常困惑。因为aducanumab在两项设计相同的3期临床试验中获得了截然相反的试验结果。是什么原因导致了aducanumab在两项试验中的表现大相径庭?渤健在10月23日公布的数据并没有给出令人信服的解释。
今日,渤健在第12届阿尔茨海默病临床试验(12th Clinical Trials on Alzheimer’s Disease, CTAD)会议上公布了大家期盼已久,万众瞩目的aducanumab在EMERGE和ENGAGE临床试验中获得的详细试验结果。
EMERGE和ENGAGE临床试验结果不同是由于什么原因?
渤健在10月23日公布的数据中,最令人惊讶的恐怕就是EMERGE和ENGAGE两项理论上设计完全相同的临床试验,却得出了完全不同的结果。在EMERGE临床试验中,接受高剂量aducanumab治疗的患者组,衡量认知能力的CDR-SB评分降低22%(评分降低意味着疾病症状恶化速度减缓),而在ENGAGE临床试验中,同样接受高剂量aducanumab治疗的患者组CDR-SB评分反而升高了2%。那么这两组患者接受的治疗有什么不同么?
这要从这两项试验的临床设计谈起。参加临床试验的AD患者可以被分为两类,一类患者携带称为ApoE ε4的等位基因(ApoE ε4+),而另一类患者不携带ApoE ε4等位基因(ApoE ε4-)。编码ApoE蛋白的等位基因在人群中有三大类型,分别为ApoE ε4,ApoE ε3,和ApoE ε2。携带ApoE ε4的群体患上AD的风险比其他群体来讲显着升高。
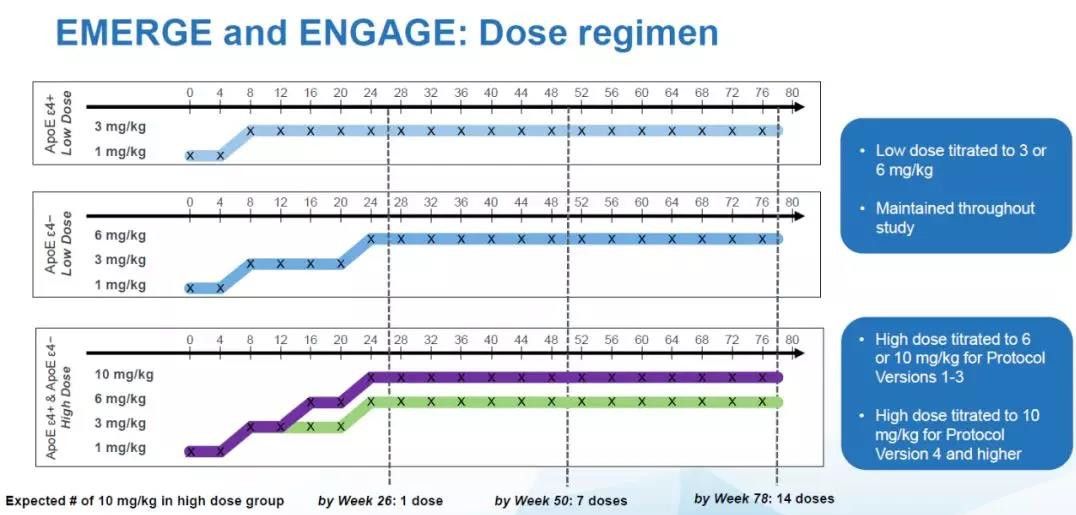
同时,渤健的前期试验结果表明,ApoE ε4+患者在接受aducanumab治疗时更容易出现一种名为ARIA的副作用。ARIA的全名是淀粉样蛋白相关成像异常(amyloid associated imaging abnormality),这可能是大脑水肿或微出血的迹象。由于这一副作用,ApoE ε4+患者在最初的试验设计中,接受的aducanumab最高剂量为6 mg/kg。而不携带ApoE ε4的患者可以接受的最高剂量为10 mg/kg。就是说,在同一个高剂量组里,根据患者是否携带ApoE ε4,他们接受的aducanumab剂量是不同的。
在2017年3月,渤健对试验流程进行了更改,根据对ARIA的研究,渤健发现这一副作用对患者的健康影响并不大,因此更新的试验流程(Protocol Version 4, Pv4)中,携带ApoE ε4的患者也可以接受最高剂量为10 mg/kg的aducanumab的治疗。 这意味着,在高剂量组中,携带ApoE ε4的患者接受的aducanumab剂量并不统一,有的患者可能接受的是6 mg/kg,而其它患者接受的是10 mg/kg。
而由于EMERGE和ENGAGE两项试验招募患者的速度不同,导致了在高剂量组中,真正接受了10 mg/kg的aducanumab治疗的患者比例有显着差别。在试验流程更改之前,在EMERGE的高剂量组中,只有21%的患者能够接受14次10 mg/kg的治疗。而在ENGAGE的高剂量组中,这一患者群的比例下降到15%。这是这两组患者之间的一个重要不同之处。
新试验流程下入组的患者群中,EMERGE和ENGAGE试验数据相似
在新的试验流程下(Post-PV4),入组患者能够接受10 mg/kg的高剂量aducanumab治疗的比例显着升高,EMERGE组中51%的患者在整个临床试验中能够接受14次10 mg/kg的治疗,而ENGAGE组中47%的患者能够接受14次 10 mg/kg的治疗。
如果我们聚焦于这些有更大几率接受最高剂量aducanumab治疗的患者亚群,那么数据分析发现,EMERGE和ENGAGE临床试验的数据非常类似。在新试验流程下入组的患者中,EMERGE和ENGAGE试验中高剂量组患者的CDR-SB分数分别下降了30%和27%。这些数据表明,在EMERGE和ENGAGE组中,持续接受10 mg/kg剂量的aducanumab治疗的患者能够获得一致的疾病症状缓解。
众多疑问有待解决
Aducanumab的详细结果显示,如果患者能够持续接受10 mg/kg的高剂量aducanumab的治疗,那么他们认知能力的下降能够得到改善。这并不意味着患者的认知能力得到了改善,而是下降的速度减慢了。不过对于AD患者来说,这有着非常重要的意义。参与这一临床试验的研究员之一,多伦多记忆项目(Toronto Memory Program)的医学主任Sharon Cohen博士说:“这意味着对于轻度患者来说,他们有更多时间能够自理,他们能够工作、购物、旅行…对于AD患者来说,最令人担忧的就是生活无法自理,我认为,这种改善对我们的患者来说非常重要,远远超过记忆测试中评分的改变数值。”
不过,即便公布了aducanumab的详细结果,仍有很多与aducanumab相关的疑问尚未得到解答。例如:
Aducanumab对不携带ApoE ε4的AD患者的疗效如何?
渤健公布的数据中并没有将患者的疗效按照是否携带ApoE ε4分类。由于Pv4的改变是让携带ApoE ε4的患者能够使用更高剂量的aducanumab,这意味着Pv4带来的正面影响可能是由于ApoE ε4阳性患者的症状改善造成的。那么高剂量的aducanumab对不携带ApoE ε4的患者是否有同样的疗效?从FDA审批的角度,这将决定aducanumab适用患者群的大小。
Aducanumab的副作用是否会影响它的批准?
ARIA是β淀粉样蛋白抗体类药物在临床试验中出现的常见副作用。在渤健公布的详细数据中,接受高剂量aducanumab治疗的患者中,~35%的患者出现ARIA相关的大脑水肿(ARIA-E),~18%到22.7%的患者出现ARIA相关的微出血(ARIA-H)。由于FDA在审评aducanumab的监管申请时将衡量它的疗效与风险比,因此ARIA事件的出现频率和严重程度也成为大家关心的问题。不过渤健表示,大多数出现ARIA的患者没有表现出明显症状,没有患者因为出现ARIA而死亡。而且随着更多这类药物临床试验的进行,医护人员现在对ARIA的控制手段更为成熟,所以它可能不再会成为限制治疗的因素。
渤健是否需要进行一项新的临床试验?
对有机会持续接受10 mg/kg剂量aducanumab治疗的患者亚组分析虽然在EMERGE和ENGAGE临床试验中表现出一致的疗效,但是这是对试验的一个亚组的事后分析。Baird公司的分析师Brian Skorney先生表示,“我们认为这一分析应该被看做一个生成假说的探索。想要真正确定(高剂量aducanumab治疗)的疗效,渤健需要进行一项新的前瞻性临床试验。”
版权声明 本网站所有注明“来源:生物谷”或“来源:bioon”的文字、图片和音视频资料,版权均属于生物谷网站所有。非经授权,任何媒体、网站或个人不得转载,否则将追究法律责任。取得书面授权转载时,须注明“来源:生物谷”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
87%用户都在用生物谷APP 随时阅读、评论、分享交流 请扫描二维码下载->
